Mass spectrometry (MS)-based proteomics is powerful for biological discovery — but the step between peptide measurements and protein-level quantities is where a surprising amount of information quietly disappears. Most workflows reduce a rich, multi-peptide signal down to a single number using simple aggregation rules. That compression has real consequences for the accuracy and reproducibility of differential expression analysis.
The Standard Approach and Its Blind Spots
Section titled “The Standard Approach and Its Blind Spots”The most common protein quantification methods — summing intensities, taking the top N peptides, or normalizing by the number of observable peptides (iBAQ) — share a common design philosophy: aggregate and move on. They treat all peptides as roughly interchangeable proxies for protein abundance, and they treat all intensity measurements as equally reliable.
In reality, neither assumption holds. Peptides from the same protein can differ dramatically in how reliably they are detected. Some are efficiently digested and ionized; others are prone to missed cleavages, suppression, or co-elution. Beyond detectability, every peptide intensity estimate carries its own uncertainty — shaped by signal-to-noise, identification confidence, and whether the peptide was observed at all in a given sample.
When these factors are ignored, the resulting protein quantity is a noisy average that obscures the underlying signal. High-confidence, well-detected peptides are given no more weight than low-confidence, barely-observed ones.
Uncertainty-Aware Quantification
Section titled “Uncertainty-Aware Quantification”Tesorai’s uncertainty-aware quantification (UAQ) approach treats each peptide measurement as what it actually is: an estimate with an associated reliability. Rather than averaging across peptides uniformly, UAQ integrates three pieces of information for every PSM:
- Intensity: the measured abundance signal
- Identification uncertainty: the confidence that the PSM is correctly assigned
- Missingness: whether and how often the peptide was detected across samples
The result is a protein-level quantity that reflects the collective evidence from all observed peptides, weighted by how much each one can be trusted.
Benchmark Results: Accuracy and Reproducibility
Section titled “Benchmark Results: Accuracy and Reproducibility”On a 3-mix species DIA Astral dataset (Van Puyvelde 2022) — where the true fold-changes between conditions are known — UAQ shows consistently lower RMSE and standard deviation in log₂ fold-change estimates compared to both Tesorai iBAQ and DIA-NN. The quantities are closer to ground truth, and more reproducible across replicates.
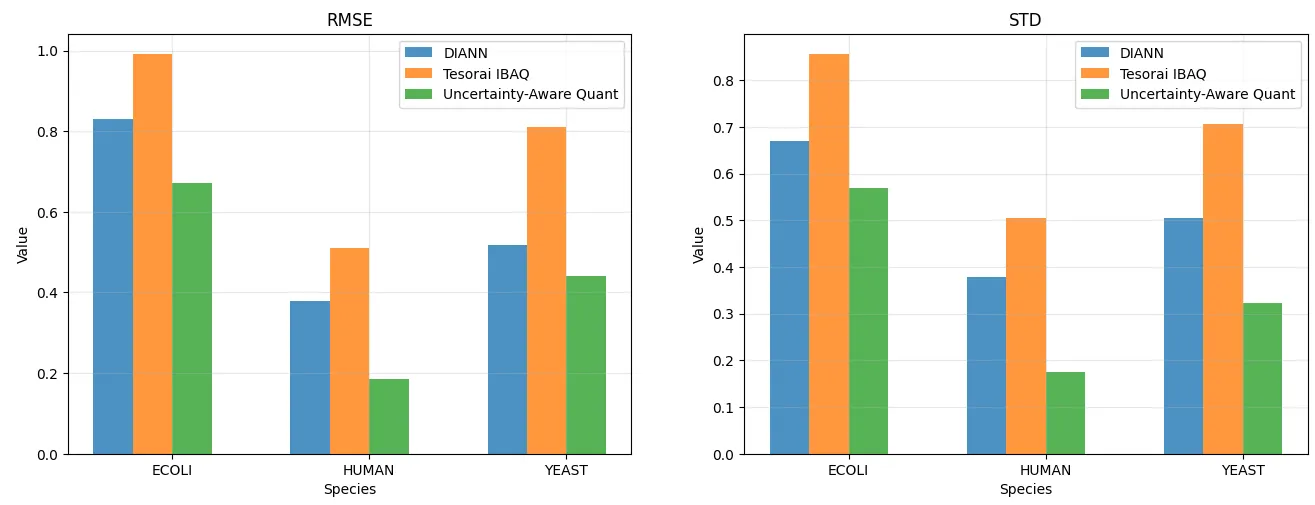
Figure 1. Improved quantification accuracy and precision with uncertainty-aware quantification Using a 3-mix species DIA Astral (Van Puyvelde 2022) dataset, we benchmark the quantification accuracy by measuring the difference between expected and observed log-2 fold-change intensities. Uncertainty-Aware Quantification method shows consistently lower RMSE and STD than Tesorai iBAQ and DIA-NN, indicating higher accuracy and reproducibility.
That accuracy gain translates directly into better differential expression results.
Fewer False Positives, More True Discoveries
Section titled “Fewer False Positives, More True Discoveries”In a quality-control experiment using human proteins mixed in equal amounts across six files, no true differential expression exists — so every detected DEP is a false positive. UAQ markedly reduces these spurious detections compared to iBAQ (p < 0.01, |log₂FC| > 1), demonstrating that better-calibrated quantities lead to more stable statistical behavior when there is no real signal.
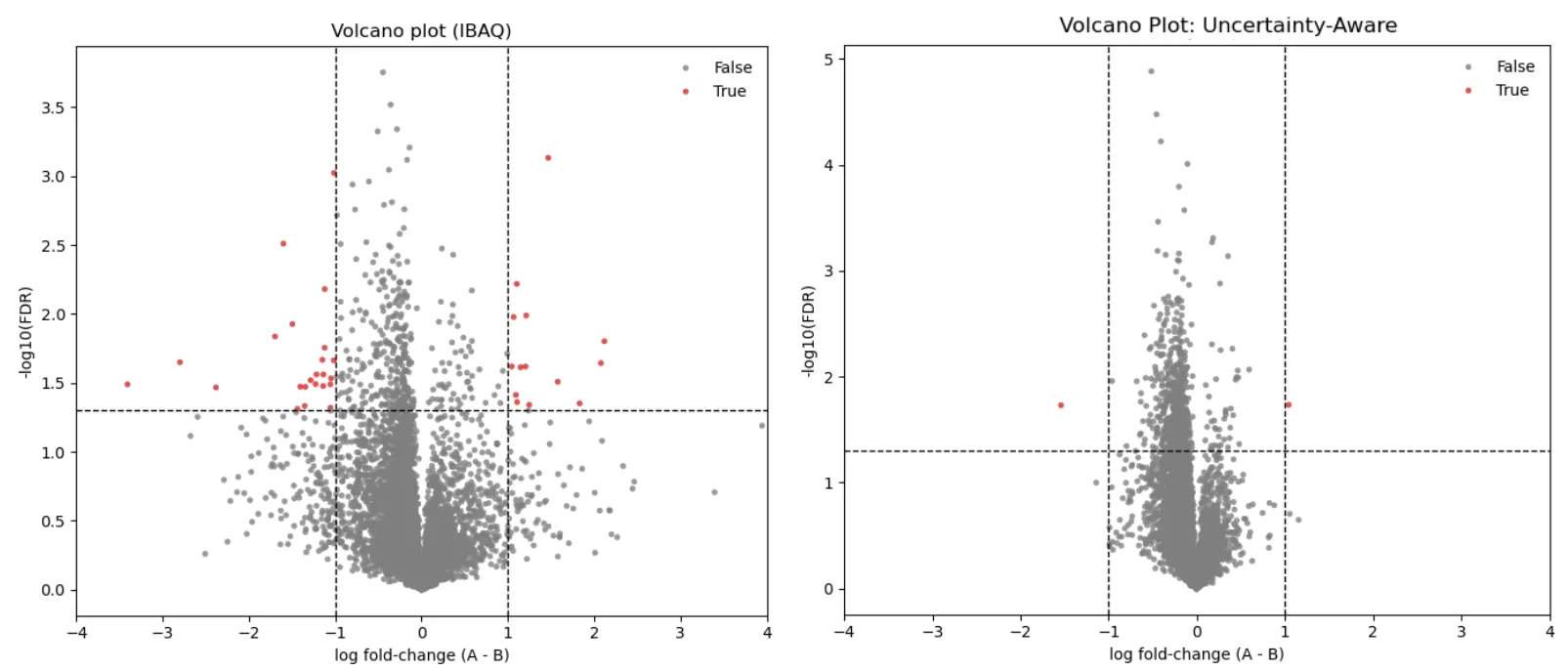
Figure 2.* Comparison of differential expression results between traditional iBAQ and the new UAQ approach in a QC dataset (Van Puyvelde 2022). We selected only human proteins, which were mixed in equal amounts across all six files. As no biological difference is expected, all detected DEPs represent false positives. UAQ markedly reduces spurious detections (p < 0.01, |log₂FC| > 1).
The complementary picture emerges in a Parkinson’s disease cohort (Zhu et al. 2024, postmortem brain tissue). Here, UAQ identifies twice as many biologically relevant DEPs as iBAQ (q < 0.02, |log₂FC| > 2):
- UAQ: NDRG2, APOA4, CTNNA2, GRIPAP1, LAMB2, PMP2, MYH11, PPFIA3
- iBAQ: NDRG2, APOA4, MAG, TMCO1
The additional proteins recovered by UAQ include several with known roles in Parkinson’s and Alzheimer’s Disease — not noise, but biologically meaningful signal that the standard approach lacks the sensitivity to detect.
Conclusion
Section titled “Conclusion”Simple aggregation methods discard information that matters. Peptide reliability, identification confidence, and missingness are not nuisances to be averaged away — they are signals about how much each measurement should be trusted. UAQ puts that information back into the quantification step, producing protein-level estimates that are more accurate, more reproducible, and more sensitive to the subtle proteomic changes that matter most in disease research.
References:
- Zhu et al. 2024. Single-cell transcriptomic and proteomic analysis of Parkinson’s disease brains. PXD030142
- Van Puyvelde 2022. A comprehensive LFQ benchmark dataset on modern day acquisition strategies in proteomics. PXD028735