Identifying peptides from mass spectrometry data is the cornerstone of proteomics. For years, the field relied on search engines using relatively simple ‘barcode matching’. While foundational, these methods often struggled to fully utilize the rich information packed within each tandem mass spectrum.
Recognizing these limitations, the community developed sophisticated tools incorporating numerous features – predicting fragment ion intensities, modeling retention times, assessing charge states, and more. Many employ powerful machine learning classifiers, often trained ‘on-the-fly’ for each specific dataset using target-decoy strategies (like the widely used Percolator). While these approaches boosted identifications, they also introduced new complexities. Relying on dozens of handcrafted features and dataset-specific training can sometimes lead to inconsistent results between runs and raises complex questions about accurate False Discovery Rate (FDR) control, especially when decoy distributions don’t perfectly mimic real targets.
We started questioning this trajectory. What if the perceived limitations of earlier methods stemmed not from a fundamental lack of information in the core data, but from incomplete utilization of the spectrum and peptide sequence context? What if a single, powerful model could learn the complex, non-linear associations directly, without needing intricate feature engineering or on-the-fly decoy training?
Introducing Tesorai Search: Deep Learning Directly on Spectra and Sequences
Section titled “Introducing Tesorai Search: Deep Learning Directly on Spectra and Sequences”This philosophy underpins Tesorai Search, our novel proteomics identification engine detailed in our latest preprint (soon to be submitted to Nature Methods!). We developed a single, large, end-to-end deep learning model. Think of it like modern large language models (indeed, it leverages similar transformer-based architectures) but trained specifically for the language of peptides and spectra.
Crucially, this model was pre-trained on a massive and diverse dataset – nearly 289 million high-quality, real peptide-spectrum matches covering various instrument types, fragmentation methods, and biological samples.
Here’s the core innovation: our model takes the entire tandem mass spectrum and the complete candidate peptide sequence as direct input. It processes this raw data end-to-end and outputs a single, precise score reflecting the likelihood of a true match.
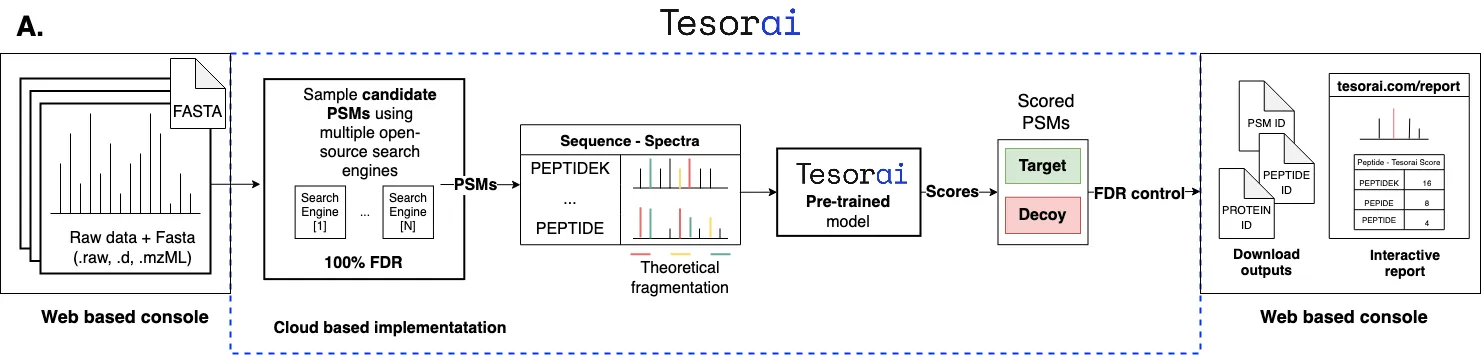
This streamlined approach deliberately avoids two common practices:
- Complex Feature Engineering: We let the model learn the relevant patterns directly from the spectrum and sequence, rather than relying on predefined, potentially limited features.
- Decoy-Based Classifier Training: Our primary scoring model is trained only on real target matches. We avoid training scoring classifiers using decoys on-the-fly for each dataset. Why? Because using decoys for training can inadvertently teach the model properties specific to the decoys themselves, potentially biasing the scores and complicating FDR estimation. We still use decoys in the standard way for final FDR estimation, but not for training the core scoring mechanism.
This simplified workflow also means fewer processing steps are required – no need for spectrum de-isotoping or score recalibration, making the process more robust and easier to manage.
Boosting Identifications Across the Board
Section titled “Boosting Identifications Across the Board”Does stripping away these complex features and on-the-fly training hurt performance? Quite the opposite. By focusing the deep learning model on the most fundamental data, Tesorai Search consistently identifies more peptides.
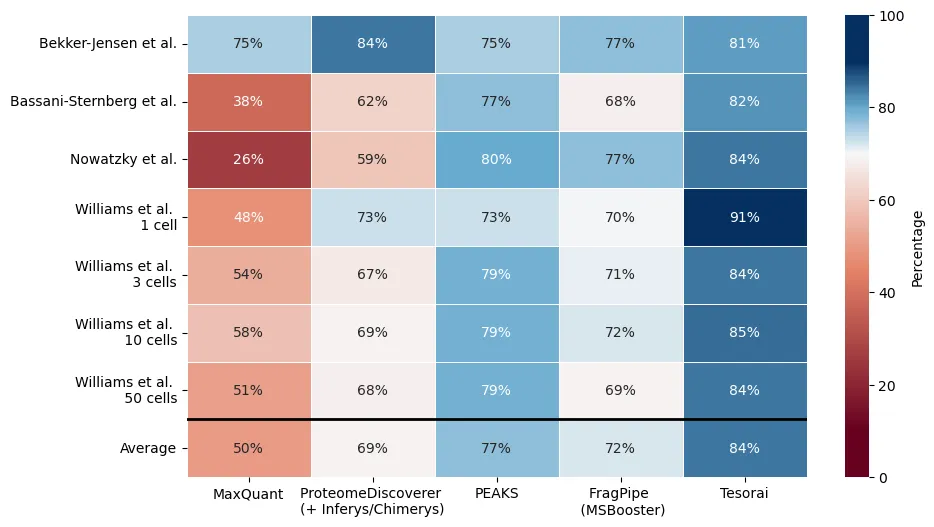
We rigorously benchmarked our engine against several leading platforms (FragPipe with MSBooster, PEAKS, ProteomeDiscoverer, MaxQuant) across a wide range of datasets: standard tryptic digests of HeLa cells, challenging low-abundance immunopeptidomics samples, and even cutting-edge low-input single-cell proteomics data.
The results demonstrate significant gains:
- Substantially More Peptide IDs: On average, Tesorai Search identified considerably more unique peptides: approximately 17% more than FragPipe, 9% more than PEAKS, 21% more than ProteomeDiscoverer, and a substantial 68% more than MaxQuant.
- Consistent High Performance: These improvements weren’t limited to one specific scenario. For instance, in immunopeptidomics datasets known for non-tryptic peptides and sparse spectra, Tesorai often yielded 15-25% more identifications than the next best tool. Similar gains were observed in single-cell analyses, pushing the boundaries of detection.
- Excellent Generalization: The model performed well even on data types it wasn’t explicitly trained on, including data from TOF instruments and spectra with TMT reporter ions.
- Reliable FDR Control: Our approach of separating the scoring model from decoy training ensures robust and accurate FDR estimation. We validated this using entrapment experiments with known non-human protein sequences spiked into human samples, confirming that our reported FDR levels are trustworthy.
Fast, Scalable, Reproducible, and Accessible Science
Section titled “Fast, Scalable, Reproducible, and Accessible Science”Performance isn’t just about IDs; it’s also about usability. We built Tesorai Search as a cloud-native platform from the ground up.
- Blazing Speed: Analyze data faster than ever. Reprocessing 250 immunopeptidomics files (a task that could take days locally) finished in under 45 minutes on our platform.
- Effortless Scalability: Handle projects of any size without worrying about local computing limitations.
- Enhanced Reproducibility: Cloud-based processing with versioned software ensures analyses can be easily reproduced by you or collaborators.
- Universal Accessibility: No complex software installations or high-performance hardware needed. Access and run Tesorai Search directly through your web browser.
The Takeaway
Section titled “The Takeaway”Tesorai Search represents a paradigm shift back towards the fundamental data in proteomics – the spectrum and the sequence. By applying a large, pre-trained deep learning model directly to this information, we achieve state-of-the-art identification sensitivity while simultaneously enhancing speed, robustness, and ease of use. We’ve shown that the full spectrum and peptide sequence are sufficient for top-tier performance, challenging the necessity for complex, multi-stage feature engineering and dataset-specific training loops.
We believe this approach not only boosts results but also makes powerful proteomics analysis more accessible and reliable. Explore the details in our preprint!